[1] 0.01801949# d(1,3)
1 - cor(x1, x3)[1] 1.866025# d(2,3)
1 - cor(x2, x3)[1] 1.755929[1] 1.773948The classification models we’ve discussed are all supervised learning techniques. The word supervised refers to the fact that we know the response variable of all of the training observations. Next up, we’ll discuss clustering which is an unsupervised technique – none of the observations have a given response variable. For example, we might want to cluster a few hundred melanoma patients based on their genetic data. We are looking for patterns in who groups together, but we don’t have a preconceived idea of which patients belong to which group.
There are also semi-supervised techniques applied to data which have some observations that are labeled and some that are not. We will not discuss semi-supervised methods in this class.
Clustering creates groups of observations via unsupervised methods. We will cover hierarchical clustering, k-means, and k-medoids. We cluster for two main reasons:
Classification – SUPERVISED! creates predictions (and prediction models) for unknown future observations via supervised methods. With classification the group membership (i.e., response variable) is known for all the training data. We covered k-NN, CART, bagging, Random Forests, and SVMs.
Many, though not all, clustering algorithms are based on distances between the objects being clustered. Mathematical properties of a distance function are the following. Consider two vectors \({\bf x}\) and \({\bf y}\) (\({\bf x}, {\bf y} \in \mathbb{R}^p\)), and the distance between them: \(d({\bf x}, {\bf y})\).
Triangle Inequality
The key to proving the triangle inequality for most of the distances relies on the Cauchy-Schwarz inequality. \[\begin{align} {\bf x} \cdot {\bf y} &= || {\bf x} || ||{\bf y}|| \cos(\theta) \\ |{\bf x} \cdot {\bf y}| &\leq || {\bf x} || ||{\bf y}|| \end{align}\]
\[d_E({\bf x}, {\bf y}) = \sqrt{\sum_{i=1}^p (x_i - y_i)^2}\]
Distance properties all check out.
Shortcomings:
Strengths:
\[\begin{align} d_P({\bf x}, {\bf y}) &= 1 - r_P ({\bf x}, {\bf y})\\ \mbox{ or } &= 1 - |r_P ({\bf x}, {\bf y})|\\ \mbox{ or } &= 1 - (r_P ({\bf x}, {\bf y}))^2\\ \end{align}\]
Notice that Euclidean distance and Pearson correlation distance are similar if the original observations are scaled. Assume that the sample mean for \({\bf x}\) (that is, \(\frac{1}{p} \sum x_i = \overline{x} = 0\)) is zero and the sample standard deviation is 1.
\[\begin{align} r_P ({\bf x}, {\bf y}) &= \frac{\sum x_i y_i - p \ \overline{x} \ \overline{y}}{(p-1)s_x s_y}\\ &= \frac{1}{(p-1)} \sum x_i y_i\\ & \ \ & \\ d_E({\bf x}, {\bf y}) &= \sqrt{\sum(x_i - y_i)^2}\\ &= \sqrt{ \sum x_i^2 + \sum y_i^2 - 2 \sum x_i y_i}\\ d_E^2 &= 2[(p-1) - \sum x_i y_i]\\ &= 2(p-1)*[1 - r_P({\bf x}, {\bf y})] \end{align}\]
\({\bf y}=a{\bf x}\) \[\begin{align} d_P({\bf x}, {\bf y}) &= 1 - r_P ({\bf x}, {\bf y})\\ &= 1 - r_P ({\bf x}, a{\bf x})\\ &= 1 - 1 = 0 \end{align}\]
\({\bf x}=(1,1,0)\), \({\bf y} = (2,1,0)\), \({\bf z} = (1,-1,0)\) \(r_P({\bf x}, {\bf y}) = 0.87\), \(r_P({\bf x}, {\bf z}) = 0\), \(r_P({\bf y}, {\bf z}) = 0.5\)
\(d_P({\bf x}, {\bf y}) + d_P({\bf y}, {\bf z}) < d_P({\bf z}, {\bf x})\) \(\rightarrow\leftarrow\)
Regular Pearson distance
[1] 0.01801949# d(1,3)
1 - cor(x1, x3)[1] 1.866025# d(2,3)
1 - cor(x2, x3)[1] 1.755929[1] 1.773948Absolute Pearson distance
Using absolute distance doesn’t fix things.
[1] 0.01801949[1] 0.1339746[1] 0.2440711[1] 0.1519941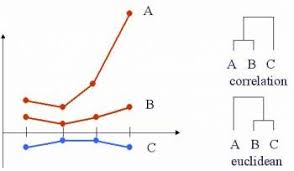
Shortcomings:
Strengths:
Spearman correlation distance uses the Spearman correlation instead of the Pearson correlation. The Spearman correlation is simply the Pearson correlation applied to the ranks of the observations. The ranking allows the Spearman distance to be resistant to outlying observations.
\[\begin{align} d_S({\bf x}, {\bf y}) &= 1 - r_S ({\bf x}, {\bf y})\\ \mbox{ or } &= 1 - |r_S ({\bf x}, {\bf y})|\\ \mbox{ or } &= 1 - (r_S ({\bf x}, {\bf y}))^2\\ \end{align}\]
Shortcomings:
Strengths:
\[\begin{align} d_C({\bf x}, {\bf y}) &= \frac{{\bf x} \cdot {\bf y}}{|| {\bf x} || ||{\bf y}||}\\ &= \frac{\sum_{i=1}^p x_i y_i}{\sqrt{\sum_{i=1}^p x_i^2 \sum_{i=1}^p y_i^2}}\\ &= 1 - r_P ({\bf x}, {\bf y}) \ \ \ \ \mbox{if } \overline{\bf x} = \overline{\bf y} = 0 \end{align}\]
Said differently,
\[\begin{align} d_P({\bf x}, {\bf y}) = d_C({\bf x} - \overline{\bf x}, {\bf y} - \overline{\bf y}) \end{align}\]
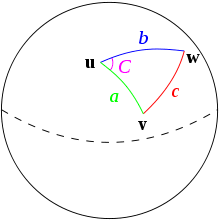
Haversine distance is the great-circle distance (i.e., the distance between two points on a sphere) which is used to measure distance between two locations on the Earth. Let \(R\) be the radius of the Earth, and (lat1,long1) and (lat2, long2) be the two locations between which to calculate a distance.
\[d_{HV} = 2 R \arcsin \sqrt{\sin^2 \bigg( \frac{lat2-lat1}{2} \bigg) + \cos(lat1) \cos(lat2) \sin^2 \bigg(\frac{long2 - long1}{2} \bigg)} \]
Shortcomings:
Strengths:
Hamming distance is the number of coordinates across two vectors whose values differ. If the vectors are binary, the Hamming distance is equivalent to the \(L_1\) norm of the difference. (Hamming distance does satisfy the properties of a distance metric.) Some methods, equivalently, calculate the proportion of coordinates that differ.
\[\begin{align} d_H({\bf x}, {\bf y}) = \sum_{i=1}^p I(x_i \ne y_i) \end{align}\]
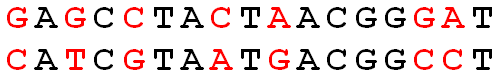
Shortcomings:
Strengths:

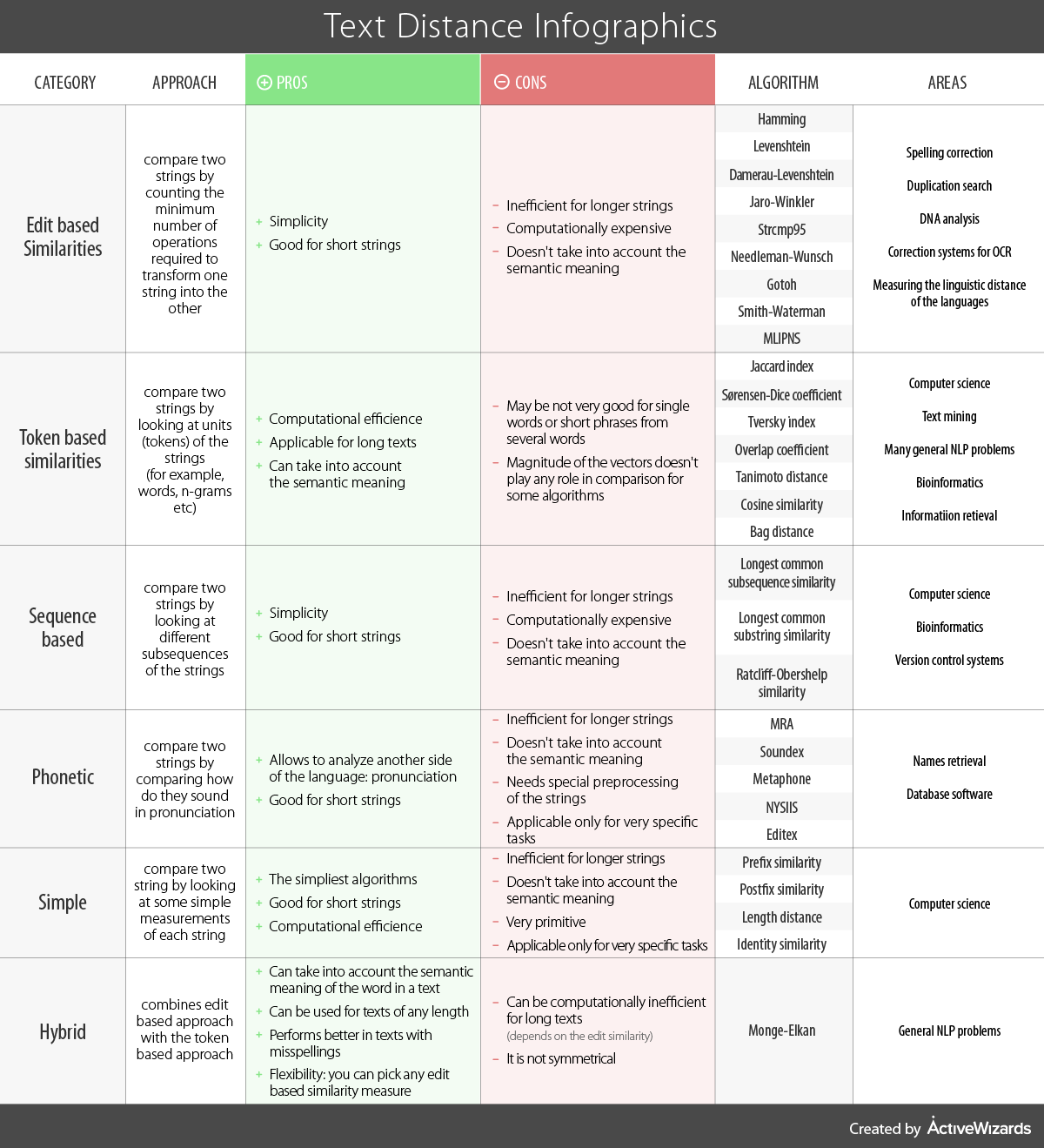
dist in R calculates the distances given above.Consider the following infographic which compares different methods for computing distances between strings.
Hierarchical Clustering is a set of nested clusters that are organized as a tree. Note that objects that belong to a child cluster also belong to the parent cluster.
Example: Consider the following images / data (from Laura Hoopes, personal communication; Molecular characterisation of soft tissue tumours: a gene expression study by Nielsen et al., The Lancet 2002). The first represents a microarray sample from aging yeast. The second is a set of 41 samples of soft-tissue tumors (columns) and a subset of 5520 genes (rows) used to characterize their molecular signatures.
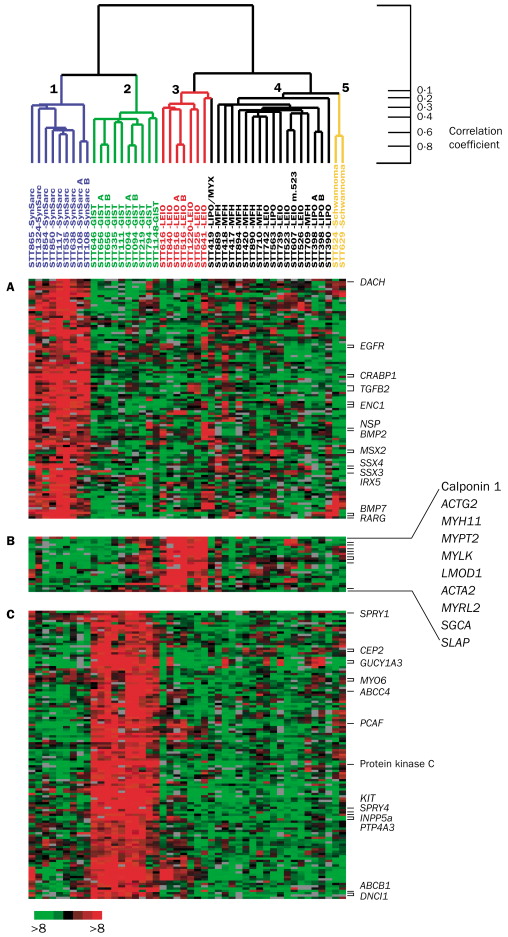
Note: the ordering of the variables (or samples) does not affect the clustering of the samples (or variables). That is: we can clustering the variables / samples either sequentially or in parallel to see trends in both relationships simultaneously. Clustering both the observations and the variables is called biclustering.
Algorithm: Agglomerative Hierarchical Clustering Algorithm
Agglomerative methods start with each object (e.g., gene) in its own group. Groups are merged until all objects are together in one group.
Divisive methods start with all objects in one group and break up the groups sequentially until all objects are individuals.
Single Linkage algorithm defines the distance between groups as that of the closest pair of individuals.
Complete Linkage algorithm defines the distance between groups as that of the farthest pair of individuals.
Average Linkage algorithm defines the distance between groups as the average of the distances between all pairs of individuals across the groups.
strengths
shortcomings
| A | B | C | D | E | |
|---|---|---|---|---|---|
| A | 0 | ||||
| B | 0.2 | 0 | |||
| C | 0.6 | 0.5 | 0 | ||
| D | 1 | 0.9 | 0.4 | 0 | |
| E | 0.9 | 0.8 | 0.5 | 0.3 | 0 |
Link A and B! \[\begin{align} d_{(AB)C} &= \min(d_{AC}, d_{BC}) = 0.5\\ d_{(AB)D} &= \min(d_{AD}, d_{BD}) = 0.9\\ d_{(AB)E} &= \min(d_{AE}, d_{BE}) = 0.8\\ \end{align}\]
| AB | C | D | E | |
|---|---|---|---|---|
| AB | 0 | |||
| C | 0.5 | 0 | ||
| D | 0.9 | 0.4 | 0 | |
| E | 0.8 | 0.5 | 0.3 | 0 |
Link D and E! \[\begin{align} d_{(AB)C} &= 0.5\\ d_{(AB)(DE)} &= \min(d_{AD}, d_{BD}, d_{AE}, d_{BE}) = 0.8\\ d_{(DE)C} &= \min(d_{CD}, d_{CE}) = 0.4\\ \end{align}\]
| AB | C | DE | |
|---|---|---|---|
| AB | 0 | ||
| C | 0.5 | 0 | |
| DE | 0.8 | 0.4 | 0 |
Link C with (DE)! \[\begin{align} d_{(AB)(CDE)} = d_{BC} = 0.5 \end{align}\]

To predict the cluster assignment for a new observation, we find the closest cluster. How we measure “closeness” is dependent on the specified type of linkage in the model:
single linkage: The new observation is assigned to the same cluster as its nearest observation from the training data.
complete linkage: The new observation is assigned to the cluster with the smallest maximum dissimilarities between training observations and the new observation.
average linkage: The new observation is assigned to the cluster with the smallest average dissimilarities between training observations and the new observation.
centroid method: The new observation is assigned to the cluster with the closest centroid, as in prediction for k_means.
Ward’s method: The new observation is assigned to the cluster with the smallest increase in error sum of squares (ESS) due to the new addition. The ESS is computed as the sum of squared Euclidean distances between observations in a cluster, and the centroid of the cluster.
It’s important to note that there is no guarantee that predict() on the training data will produce the same results as extract_cluster_assignments(). The process by which clusters are created during the agglomerations results in a particular partition; but if a training observation is treated as new data, it is predicted in the same manner as truly new information.
Consider a silly example using complete linkage:
\[x_1 = 1, x_2 = 2, x_3 = 5, x_4 = 9, x_5 = 10, x_6 = -3\]
Now consider a new observation, \(x^* = x_3 = 5\). Using complete linkage, \(x^*\) is closer to \(\{x_4, x_5 \}\) (distance = 5) than to \(\{x_1, x_2, x_3, x_6 \}\) (distance = 8).
Again, using the penguins dataset, hierarchical clustering will be run. We can look at the dendrogram, in particular its alignment to the categorical variables like species and island.
Notice that we are only using the numerical variables. And the numerical variables have been scaled (subtract the mean and divide by the standard deviation).
Full example has been adapted from https://tidyclust.tidymodels.org/articles/hier_clust.html. You’ll need the tidyclust R package.
penguins <- penguins |>
select(bill_length_mm, bill_depth_mm,
flipper_length_mm, body_mass_g,
island, species) |>
drop_na()hc_rec <- recipe(~ ., data = penguins) |>
update_role(island, new_role = "ID") |>
update_role(species, new_role = "ID") |>
step_rm(all_nominal()) |>
step_normalize(all_predictors())hc_model <- hier_clust(
num_clusters = 3,
linkage_method = "average") |>
set_engine("stats")
hc_modelHierarchical Clustering Specification (partition)
Main Arguments:
num_clusters = 3
linkage_method = average
Computational engine: stats hc_work <- workflow() |>
add_recipe(hc_rec) |>
add_model(hc_model)
hc_work══ Workflow ════════════════════════════════════════════════════════════════════
Preprocessor: Recipe
Model: hier_clust()
── Preprocessor ────────────────────────────────────────────────────────────────
2 Recipe Steps
• step_rm()
• step_normalize()
── Model ───────────────────────────────────────────────────────────────────────
Hierarchical Clustering Specification (partition)
Main Arguments:
num_clusters = 3
linkage_method = average
Computational engine: stats hc_fit <- hc_work |>
fit(data = penguins)
hc_fit |>
summary() Length Class Mode
pre 3 stage_pre list
fit 2 stage_fit list
post 1 stage_post list
trained 1 -none- logicalhc_fit |> extract_fit_summary() |> str()List of 7
$ cluster_names : Factor w/ 3 levels "Cluster_1","Cluster_2",..: 1 2 3
$ centroids : tibble [3 × 4] (S3: tbl_df/tbl/data.frame)
..$ bill_length_mm : num [1:3] -0.369 0.603 2.253
..$ bill_depth_mm : num [1:3] 0.617 -1.123 -0.368
..$ flipper_length_mm: num [1:3] -0.65 1.13 2.05
..$ body_mass_g : num [1:3] -0.612 1.06 1.977
$ n_members : int [1:3] 219 119 4
$ sse_within_total_total: num [1:3] 283.19 111.49 2.01
$ sse_total : num 656
$ orig_labels : NULL
$ cluster_assignments : Factor w/ 3 levels "Cluster_1","Cluster_2",..: 1 1 1 1 1 1 1 1 1 1 ...hc_fit |> extract_centroids()# A tibble: 3 × 5
.cluster bill_length_mm bill_depth_mm flipper_length_mm body_mass_g
<fct> <dbl> <dbl> <dbl> <dbl>
1 Cluster_1 -0.369 0.617 -0.650 -0.612
2 Cluster_2 0.603 -1.12 1.13 1.06
3 Cluster_3 2.25 -0.368 2.05 1.98 We can use the model to predict in which cluster each penguin belongs. Note that the predicted cluster
hc_preds <- hc_fit |> predict(penguins)
hc_preds# A tibble: 342 × 1
.pred_cluster
<fct>
1 Cluster_1
2 Cluster_1
3 Cluster_1
4 Cluster_1
5 Cluster_1
6 Cluster_1
7 Cluster_1
8 Cluster_1
9 Cluster_1
10 Cluster_1
# ℹ 332 more rowsbind_cols(hc_preds,
extract_cluster_assignment(hc_fit) ) |>
select(.pred_cluster, .cluster) |>
table() .cluster
.pred_cluster Cluster_1 Cluster_2 Cluster_3
Cluster_1 218 0 0
Cluster_2 0 99 0
Cluster_3 1 20 4The basic dendrogram:
We have to get the hclust “object” from the fit, and transform it to make the dendrogram.
library(ggdendro)
penguin_hclust <- hc_fit |>
extract_fit_engine()
penguin_dend <- penguin_hclust |>
as.dendrogram()
ggdendro::ggdendrogram(penguin_dend)
Zooming in on a cluster:
ggdendrogram(penguin_dend[[1]])
ggdendrogram(penguin_dend[[2]])
ggdendrogram(penguin_dend[[1]][[1]])
Adding color to branches and nodes
library(colorspace) # get nice colors
species_col <- rev(rainbow_hcl(3))[as.numeric(penguins$species)]library(dendextend)
# order the branches as closely as possible to the original order
penguin_dend <- rotate(penguin_dend, 1:342)We don’t usually have a way to check (i.e., “unsupervised”)!
penguins |>
drop_na(bill_length_mm, bill_depth_mm, flipper_length_mm, body_mass_g) |>
select(island) |>
cbind(cluster = cutree(penguin_hclust, k = 3) ) |>
table() cluster
island 1 2 3
Biscoe 44 119 4
Dream 124 0 0
Torgersen 51 0 0penguins |>
drop_na(bill_length_mm, bill_depth_mm, flipper_length_mm, body_mass_g) |>
select(species) |>
cbind(cluster = cutree(penguin_hclust, k = 3) ) |>
table() cluster
species 1 2 3
Adelie 151 0 0
Chinstrap 68 0 0
Gentoo 0 119 4Partition Clustering is a division of the set of data objects into \(K\) non-overlapping subsets (clusters) with each observation falling into exactly one cluster.
In contrast to hierarchical clustering where results were given for any (and all!) number of clusters, partitioning methods typically start with a given \(k\) value and a set of distances. The goal is to partition the observations into \(k\) groups such that an objective function is optimized. The number of possible partitions is roughly to \(n^k / k!\) (note: \(100^{5} / 5! = 83\) million). [The exact number can be computed using Sterling numbers.] So instead of looking through all of the partitions, we step through a recursive algorithm.
A fun applet!!
https://www.naftaliharris.com/blog/visualizing-k-means-clustering/
\(k\)-means clustering is an unsupervised partitioning algorithm designed to find a partition of the observations such that the following objective function is minimized (find the smallest within cluster sum of squares):
\[\text{arg}\,\min\limits_{C_1, \ldots, C_k} \Bigg\{ \sum_{k=1}^K 2 \sum_{i \in C_k} \sum_{j=1}^p (x_{ij} - \overline{x}_{kj})^2 \Bigg\}\]
As described in the algorithm below, reallocating observations can only improve the minimization criteria with the algorithm stopping when no changes of the observations will lower the objective function. The algorithm leads to a local optimum, with no confirmation that the global minimum has occurred. Often the \(k\)- means algorithm is run multiple times with different random starts, and the partition leading to the lowest objective criteria is chosen.
Note that the following algorithm is simply one \(k\)-means algorithm. Other algorithms could include a different way to set the starting values, a different decision on when to recalculate the centers, what to do with ties, etc.
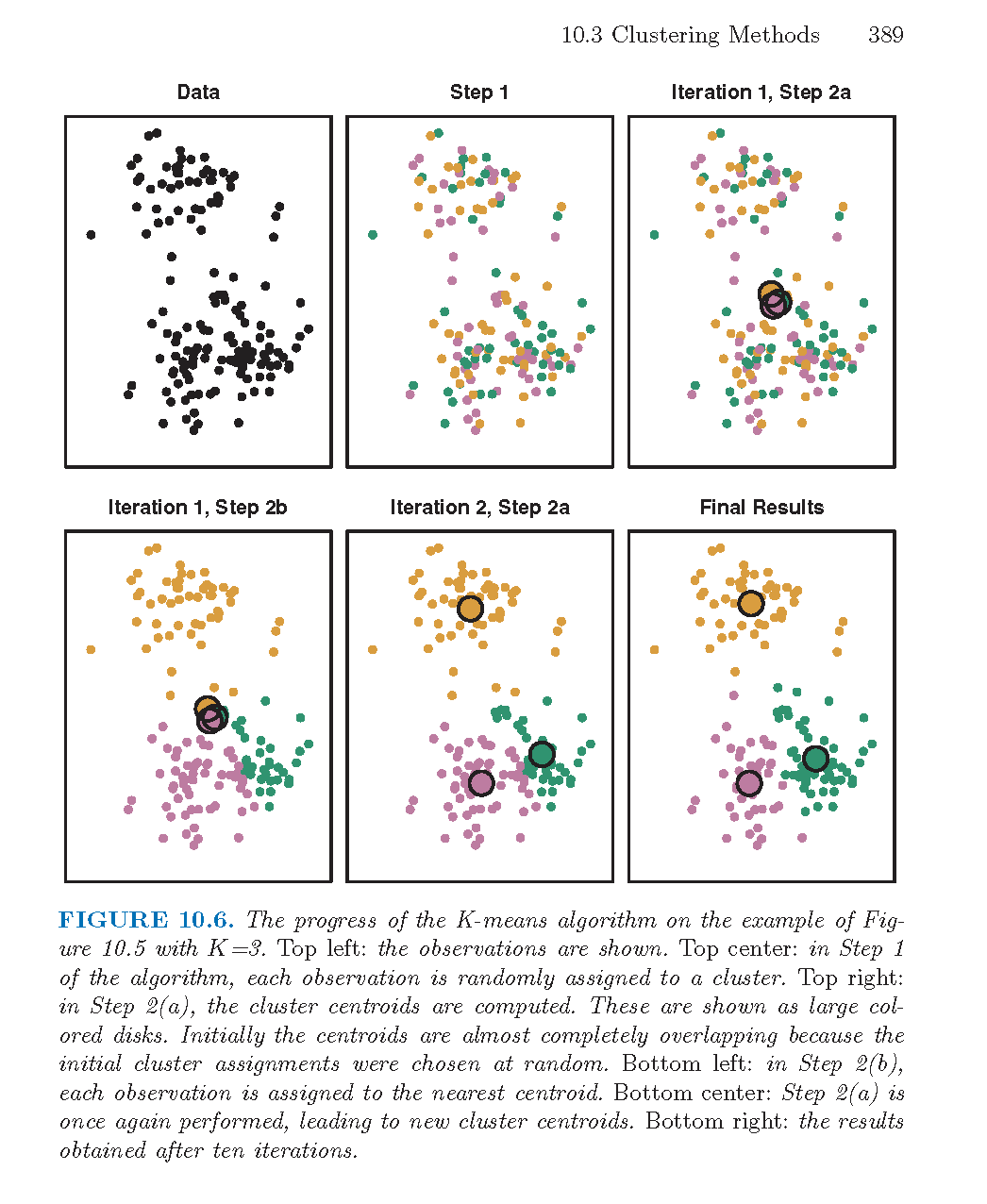
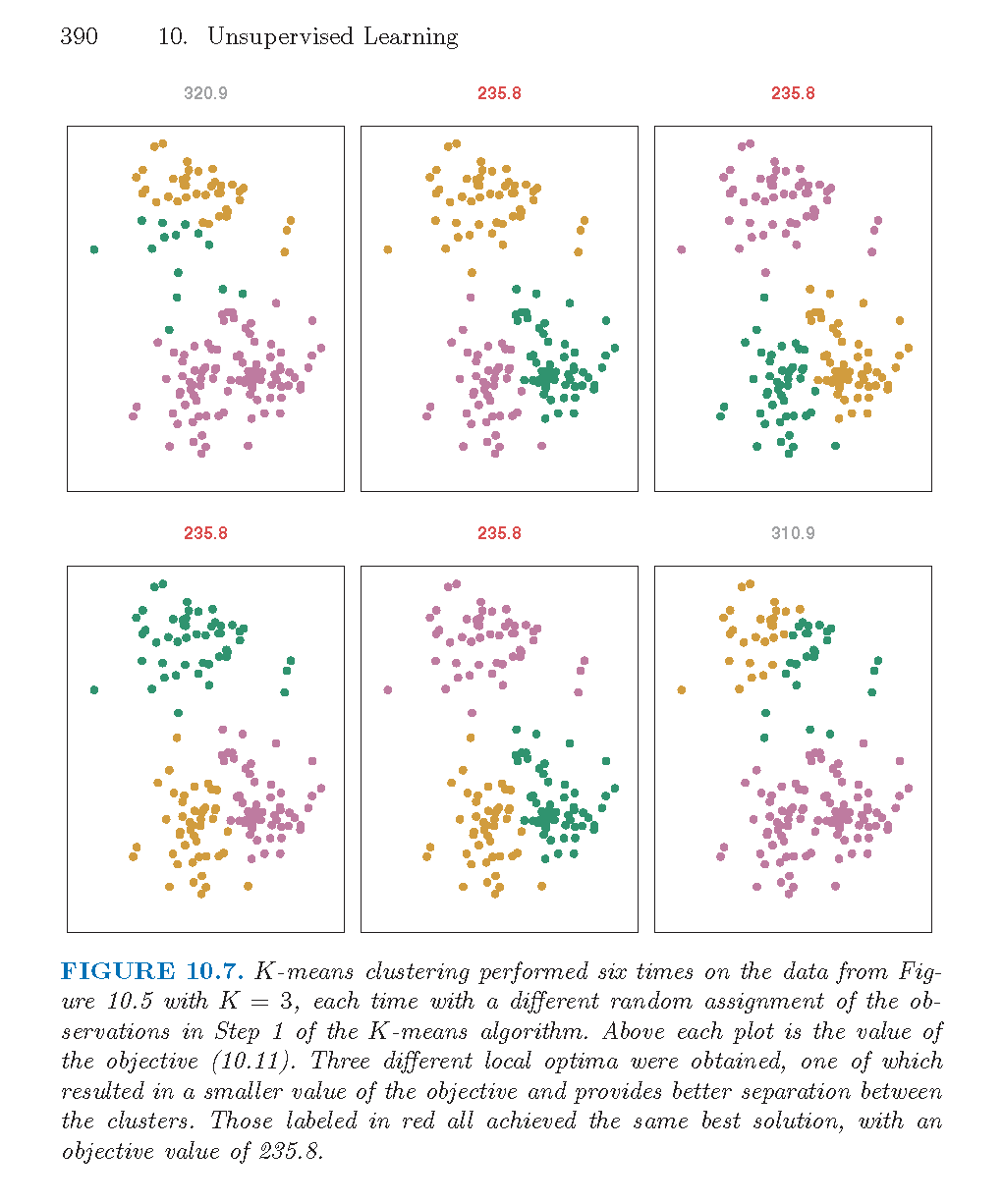
Algorithm: \(k\)-Means Clustering
Why does the \(k\)-means algorithm converge / (local) minimize the objective function?
Note that if the variables are on very different scales, whichever variable is larger in magnitude will dominate the distances (and therefore the clustering). Unless you explicitly want that to happen (which would be odd), you should scale the variables (subtract the mean and divide by the standard deviation) so that the distance is calculated on Z-scores instead of on the raw data.
In the example below, the \(k=2\) k-means algorithm is not able to see the cigar-shaped structure (on the raw data) because the distances are dominated by the x1 variable (which does not differentiate the clusters).
set.seed(47)
norm_clust <- data.frame(
x1 = rnorm(1000, 0, 15),
x2 = c(rnorm(500, 5, 1), rnorm(500, 0, 1)))
norm_clust |>
kmeans(centers = 2) |>
augment(norm_clust) |>
ggplot() +
geom_point(aes(x = x1, y = x2, color = .cluster)) +
ggtitle("k-means (k=2) on raw data")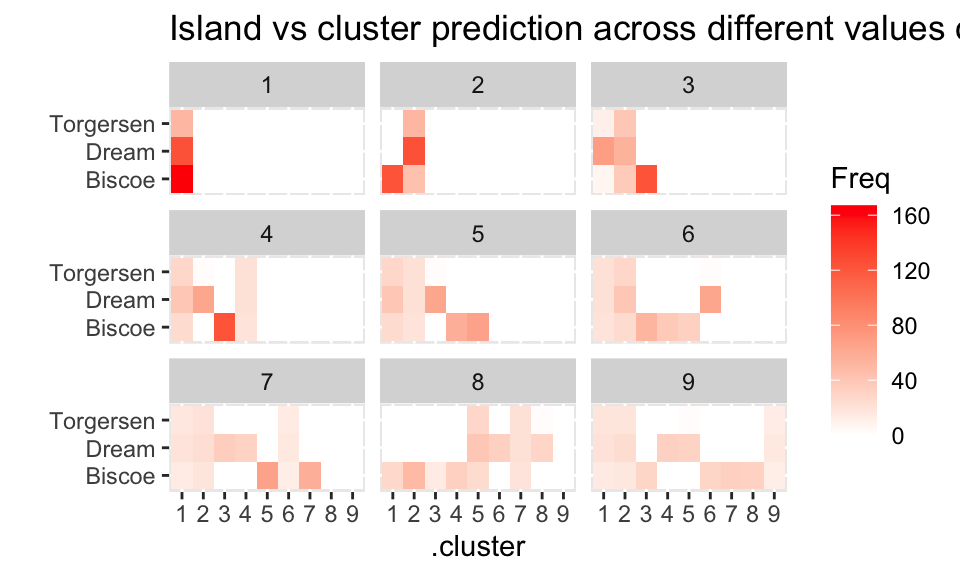
norm_clust |>
mutate(across(everything(), scale)) |>
kmeans(centers = 2) |>
augment(norm_clust) |>
ggplot() +
geom_point(aes(x = x1, y = x2, color = .cluster)) +
ggtitle("k-means (k=2) on normalized / scaled data")
strengths
shortcomings
Again, using the penguins dataset, \(k\)-means clustering will be run. We try multiple different values of \(k\) to come up with a partition of the penguins. We can look at the clusterings on scatterplots of the numerical variables. We can also check to see if the clustering aligns with the categorical variables like species and island.
Notice that we are only using the numerical variables. And the numerical variables have been scaled (subtract the mean and divide by the standard deviation).
Full example has been adapted from https://tidyclust.tidymodels.org/articles/k_means.html.
kmean_rec <- recipe(~ ., data = penguins) |>
step_naomit(all_predictors()) |>
update_role(island, new_role = "ID") |>
update_role(species, new_role = "ID") |>
update_role(sex, new_role = "ID") |>
step_rm(all_nominal()) |>
step_normalize(all_predictors())stats engine lets us tidy() and glance() and augment(). With the ClusterR engine, we can only augment() (see below).
kmean_model <- k_means(
num_clusters = 3) |>
set_engine("stats")
# set_engine("ClusterR", initializer = "random") # need the ClusterR package
kmean_modelK Means Cluster Specification (partition)
Main Arguments:
num_clusters = 3
Computational engine: stats kmean_work <- workflow() |>
add_recipe(kmean_rec) |>
add_model(kmean_model)
kmean_work══ Workflow ════════════════════════════════════════════════════════════════════
Preprocessor: Recipe
Model: k_means()
── Preprocessor ────────────────────────────────────────────────────────────────
3 Recipe Steps
• step_naomit()
• step_rm()
• step_normalize()
── Model ───────────────────────────────────────────────────────────────────────
K Means Cluster Specification (partition)
Main Arguments:
num_clusters = 3
Computational engine: stats kmean_fit <- kmean_work |>
fit(data = penguins)
kmean_fit |>
summary() Length Class Mode
pre 3 stage_pre list
fit 2 stage_fit list
post 1 stage_post list
trained 1 -none- logicalkmean_summary <- kmean_fit |> extract_fit_summary()
kmean_summary |> str()List of 7
$ cluster_names : Factor w/ 3 levels "Cluster_1","Cluster_2",..: 1 2 3
$ centroids : tibble [3 × 5] (S3: tbl_df/tbl/data.frame)
..$ bill_length_mm : num [1:3] -1.046 0.621 0.656
..$ bill_depth_mm : num [1:3] 0.474 0.825 -1.098
..$ flipper_length_mm: num [1:3] -0.911 -0.268 1.157
..$ body_mass_g : num [1:3] -0.787 -0.357 1.09
..$ year : num [1:3] -0.0923 0.0605 0.0538
$ n_members : int [1:3] 130 89 123
$ sse_within_total_total: num [1:3] 252 209 257
$ sse_total : num 1705
$ orig_labels : int [1:342] 1 1 1 1 1 1 1 1 2 1 ...
$ cluster_assignments : Factor w/ 3 levels "Cluster_1","Cluster_2",..: 1 1 1 1 1 1 1 1 2 1 ...kmean_fit |> extract_centroids()# A tibble: 3 × 6
.cluster bill_length_mm bill_depth_mm flipper_length_mm body_mass_g year
<fct> <dbl> <dbl> <dbl> <dbl> <dbl>
1 Cluster_1 -1.05 0.474 -0.911 -0.787 -0.0923
2 Cluster_2 0.621 0.825 -0.268 -0.357 0.0605
3 Cluster_3 0.656 -1.10 1.16 1.09 0.0538The function augment() works at the per-observation level:
kmean_fit |> augment(new_data = penguins |> drop_na())# A tibble: 333 × 9
.pred_cluster species island bill_length_mm bill_depth_mm flipper_length_mm
<fct> <fct> <fct> <dbl> <dbl> <int>
1 Cluster_1 Adelie Torgers… 39.1 18.7 181
2 Cluster_1 Adelie Torgers… 39.5 17.4 186
3 Cluster_1 Adelie Torgers… 40.3 18 195
4 Cluster_1 Adelie Torgers… 36.7 19.3 193
5 Cluster_1 Adelie Torgers… 39.3 20.6 190
6 Cluster_1 Adelie Torgers… 38.9 17.8 181
7 Cluster_1 Adelie Torgers… 39.2 19.6 195
8 Cluster_1 Adelie Torgers… 41.1 17.6 182
9 Cluster_1 Adelie Torgers… 38.6 21.2 191
10 Cluster_1 Adelie Torgers… 34.6 21.1 198
# ℹ 323 more rows
# ℹ 3 more variables: body_mass_g <int>, sex <fct>, year <int>The function tidy() works at the per-cluster level:
kmean_fit |> tidy()# A tibble: 3 × 8
bill_length_mm bill_depth_mm flipper_length_mm body_mass_g year size
<dbl> <dbl> <dbl> <dbl> <dbl> <int>
1 -1.05 0.474 -0.911 -0.787 -0.0923 130
2 0.621 0.825 -0.268 -0.357 0.0605 89
3 0.656 -1.10 1.16 1.09 0.0538 123
# ℹ 2 more variables: withinss <dbl>, cluster <fct>The function glance() works at the per-model level:
kmean_fit |> glance()# A tibble: 1 × 4
totss tot.withinss betweenss iter
<dbl> <dbl> <dbl> <int>
1 1705. 718. 987. 2k_func <- function(k = 3, mydata = penguins) {
workflow() |>
add_recipe(recipe(~ ., data = mydata) |> # recipe
step_naomit(all_predictors()) |>
update_role(island, new_role = "ID") |>
update_role(species, new_role = "ID") |>
update_role(sex, new_role = "ID") |>
step_rm(all_nominal()) |>
step_normalize(all_predictors())) |>
add_model(k_means(num_clusters = k) |> # model
set_engine("stats")) |>
fit(data = mydata |> drop_na()) # fit
} kmax <- 9
penguin_kclusts <-
tibble(k = 1:kmax) |>
mutate(
penguin_kclust = map(k, k_func, mydata = penguins |> drop_na()),
tidied = map(penguin_kclust, tidy),
glanced = map(penguin_kclust, glance),
augmented = map(penguin_kclust, augment, penguins |> drop_na())
)
penguin_kclusts# A tibble: 9 × 5
k penguin_kclust tidied glanced augmented
<int> <list> <list> <list> <list>
1 1 <workflow> <tibble [1 × 8]> <tibble [1 × 4]> <tibble [333 × 9]>
2 2 <workflow> <tibble [2 × 8]> <tibble [1 × 4]> <tibble [333 × 9]>
3 3 <workflow> <tibble [3 × 8]> <tibble [1 × 4]> <tibble [333 × 9]>
4 4 <workflow> <tibble [4 × 8]> <tibble [1 × 4]> <tibble [333 × 9]>
5 5 <workflow> <tibble [5 × 8]> <tibble [1 × 4]> <tibble [333 × 9]>
6 6 <workflow> <tibble [6 × 8]> <tibble [1 × 4]> <tibble [333 × 9]>
7 7 <workflow> <tibble [7 × 8]> <tibble [1 × 4]> <tibble [333 × 9]>
8 8 <workflow> <tibble [8 × 8]> <tibble [1 × 4]> <tibble [333 × 9]>
9 9 <workflow> <tibble [9 × 8]> <tibble [1 × 4]> <tibble [333 × 9]>For each of the values of \(k\) the augmented, tidyed, and glanced information can be calculated. Note that you might want to find how the total within sum of squares decreases as a function of \(k\). (Will the within sum of squares always decrease as a function of \(k?\) Does it here? Why not?) It seems as though \(k=3\) or \(k=4\) is probably sufficient (larger values of \(k\) do not reduce the sums of squares substantially).
clusterings |>
ggplot(aes(x = k, y = tot.withinss)) +
geom_line() +
geom_point() + ylab("") +
ggtitle("Total Within Sum of Squares")
From the two variables flipper_length_mm and bill_lengh_mm it seems that two clusters is probably sufficient. However, it would probably make sense to look at the cluster colorings across all four of the variables and in higher dimensional space (e.g., 3-D projections).
assignments |>
ggplot(aes(x = flipper_length_mm, y = bill_length_mm)) +
geom_point(aes(color = .pred_cluster), alpha = 0.8) +
facet_wrap(~ k)
Based on species, the clustering (\(k=2\)) seems to separate out Gentoo, but it can’t really differentiate Adelie and Chinstrap. If we let \(k=4\), we get an almost perfect partition of the three species.
species
.pred_cluster Adelie Chinstrap Gentoo
Cluster_1 138 7 0
Cluster_2 8 61 0
Cluster_3 0 0 60
Cluster_4 0 0 59
Cluster_5 0 0 0
Cluster_6 0 0 0
Cluster_7 0 0 0
Cluster_8 0 0 0
Cluster_9 0 0 0assignments |>
group_by(k) |>
select(.pred_cluster, species) |>
table() |>
as.data.frame() |>
ggplot() +
geom_tile(aes(x = .pred_cluster, y = species, fill = Freq)) +
facet_wrap( ~ k) + ylab("") +
scale_fill_gradient(low = "white", high = "red") +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust = 1)) +
ggtitle("Species vs cluster prediction across different values of k")
For island, it seems like the clustering isn’t able (really, at all!) to distinguish the penguins from the three islands.
island
.pred_cluster Biscoe Dream Torgersen
Cluster_1 42 60 43
Cluster_2 2 63 4
Cluster_3 60 0 0
Cluster_4 59 0 0
Cluster_5 0 0 0
Cluster_6 0 0 0
Cluster_7 0 0 0
Cluster_8 0 0 0
Cluster_9 0 0 0assignments |>
group_by(k) |>
select(.pred_cluster, island) |>
table() |>
as.data.frame() |>
ggplot() +
geom_tile(aes(x = .pred_cluster, y = island, fill = Freq)) +
facet_wrap( ~ k) + ylab("") +
scale_fill_gradient(low = "white", high = "red") +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust = 1)) +
ggtitle("Island vs cluster prediction across different values of k")
As an alternative to \(k\)-means, Kaufman and Rousseeuw developed Partitioning around Medoids (Finding Groups in Data: an introduction to cluster analysis, 1990). The particular strength of PAM is that it allows for any dissimilarity metric. That is, a dissimilarity based on correlations is no problem, but the algorithm gets more complicated because the “center” is no longer defined in Euclidean terms.
The two main steps are to Build (akin to assigning points to clusters) and to Swap (akin to redefining cluster centers). The objective function the algorithm tries to minimize is the average dissimilarity of objects to the closest representative object. (The PAM algorithm is a good (not necessarily global optimum) solution to minimizing the objective function.)
\[\text{arg}\,\min\limits_{C_1, \ldots, C_k} \Bigg\{ \sum_{k=1}^K \sum_{i \in C_k}D_i \Bigg\} = \text{arg}\,\min\limits_{C_1, \ldots, C_k} \Bigg\{ \sum_{k=1}^K \sum_{i \in C_k}d(x_i, m_k) \Bigg\}\] Where \(D_i\) represents this distance from observation \(i\) to the closest medoid, \(m_k\).
strengths
shortcomings
Algorithm: Partitioning Around Medoids (From The Elements of Statistical Learning (2001), by Hastie, Tibshirani, and Friedman, pg 469.)
Randomly assign a number, from 1 to \(K\), to each of the observations. These serve as initial cluster assignments for the observations.
Iterate until the cluster assignments stop changing:
Consider observation \(i \in\) cluster \(Clus1\). Let \[\begin{align} d(i, Clus2) &= \mbox{average dissimilarity of } i \mbox{ to all objects in cluster } Clus2\\ a(i) &= \mbox{average dissimilarity of } i \mbox{ to all objects in } Clus1.\\ b(i) &= \min_{Clus2 \ne Clus1} d(i,Clus2) = \mbox{distance to the next closest neighbor cluster}\\ \mbox{ silhouette width} &= s(i) = \frac{b(i) - a(i)}{\max \{ a(i), b(i) \}}\\ & &\\ \mbox{average}_{i \in Clus1} s(i) &= \mbox{average silhouette width for cluster $Clus1$} \end{align}\] Note that if \(a(i) < b(i)\) then \(i\) is well classified with a maximum \(s(i) = 1\). If \(a(i) > b(i)\) then \(i\) is not well classified with a minimum \(s(i) = -1\).
The goal is to have a large silhouette width.
Diameter of cluster \(Clus1\) (within cluster measure) \[\begin{align} \mbox{diameter} = \max_{i,j \in Clus1} d(i,j) \end{align}\]
Separation of cluster \(Clus1\) (between cluster measure) \[\begin{align} \mbox{separation} = \min_{i \in Clus1, j \notin Clus1} d(i,j) \end{align}\]
\(L^*\): a cluster with diameter \(<\) separation; \(L\): a cluster with \(\max_{j \in Clus1} d(i,j) < \min_{k \notin Clus1} d(i,k)\).
| A | B | C | D | E | |
|---|---|---|---|---|---|
| A | 0 | ||||
| B | 0.2 | 0 | |||
| C | 0.6 | 0.5 | 0 | ||
| D | 1 | 0.9 | 0.4 | 0 | |
| E | 0.9 | 0.8 | 5 | 0.3 | 0 |
Start by considering the random allocation (AC) and (BDE)
As a second step, calculate the within cluster sums of distances:
A: 0.6
C: 0.6
B: 0.9 + 0.8 = 1.7
D: 0.9 + 0.3 = 1.2
E: 0.8 + 0.3 = 1.1
For cluster 1, it doesn’t matter if we choose A or C (let’s choose C). For cluster 2, we should choose E (it is the most “central” as measured by its closer distance to both B and D).
Cluster1: C and all the points that are closer to C than E. A and B are both closer to C than to E. Cluster1 will be (A,B,C).
Cluster2: E and all the points that are closer to E than C. Only D is closer to E than C. Cluster2 will be (D,E)
A: 0.2 + 0.6 = 0.8
B: 0.2 + 0.5 = 0.7
C: 0.6 + 0.5 = 1.1
D: 0.3
E: 0.3
Cluster1 now has a medoid of B. Cluster2 (we choose randomly) has a medoid of D.
Cluster1: B and A (A,B)
Cluster2: D and C, E (D, C, E)
| A | B | C | D | |
|---|---|---|---|---|
| A | 0 | |||
| B | 0.2 | 0 | ||
| C | 0.6 | 0.5 | 0 | |
| D | 1 | 0.9 | 0.4 | 0 |
(Note: this is the same matrix as before, but with only 4 observations.)
Consider the data with (AB)(CD) as the clusters, we can calculate the previous metrics:
is based on a confusion matrix comparing either a known truth (labels) or comparing two different clusterings (e.g., comparing \(k\)-means and hierarchical clustering). Let the two different clusterings be called partition1 and partition2. * a is the number of pairs of observations put together in both partition1 and partition2 * b is the number of pairs of observations together in partition1 and apart in partition2 * c is the number of pairs of observations together in partition2 and apart in partition1 * d is the number of pairs of observations apart in both partitions
\[\mbox{Rand index} = \frac{a+d}{a+b+c+d}\]
The cool thing about the Rand index is that the partitions don’t have to even have the same number of clusters. They can be absolutely any two clusterings (one might be known labels, for example). Details on the Adjusted Rand index are given at http://faculty.washington.edu/kayee/pca/supp.pdf (basic idea is to center and scale the Rand index so that the values are more meaningful).
LDA views each document as a mixture of a small (predefined) number of topics that describe a set of documents. Each word (typically very common and extremely rare words are removed before modeling) represents a an occurrence generated by one of the document’s topics (where the document is modeled as a mixture of topics). Through LDA, the model learns both the composition of each topic and the topic mixture of each document.
From Wikipedia:
In natural language processing, latent Dirichlet allocation (LDA) is a generative statistical model that allows sets of observations to be explained by unobserved groups that explain why some parts of the data are similar. For example, if observations are words collected into documents, it posits that each document is a mixture of a small number of topics and that each word’s creation is attributable to one of the document’s topics.
Algorithm: Latent Dirichlet Allocation
How can we figure out (unsupervised!) the underlying topic of each of a set of documents?1
Start with documents that each contain words:

As a human, assign topics: Science, Politics, Sports. Here is one potential assignment:

What if you don’t have any idea what the words mean (i.e., what if you are the computer)? It would be much harder to group the words!

without using the definitions of the words, assign generic topics: Topic 1 , Topic 2, Topic 3


but let’s use colors to make it easier to track…



Color each word with red , green, blue such that
Organize the words one at a time, trying to make the articles (Goal #1) and words (Goal #2) as consistent as possible.
\(\Rightarrow\) assume that all the other words are correct, and try to predict the topic of a given word.

Do we color ball red , green, or blue?
| Topic 1 | Topic 2 | Topic 3 |
|---|---|---|
| Doc A: how many words in Topic 1 | Doc A: how many words in Topic 2 | Doc A: how many words in Topic 3 |
| 2 | 0 | 2 |

| Topic 1 | Topic 2 | Topic 3 |
|---|---|---|
| How often is ball in Topic 1 | How often is ball in Topic 2 | How often is ball in Topic 3 |
| 3 | 1 | 0 |
\[ \begin{align}P(z_i = k' | z_{-i}, W) \propto & \frac{\# \mbox{ words in $d_i$ with $k'$} + \alpha}{\# \mbox{ words in $d_i$ with any topic} + K \cdot \alpha} \cdot \\ &\frac{\# \mbox{ times $i^{th}$ word is in topic $k'$} + \beta}{\# \mbox{ words in topic $k'$} + V \cdot \beta} \end{align} \]
\[ \begin{align}P(ball_1 = \mbox{Topic 1} | z_{-i}, W) \propto& \frac{2 + \alpha}{4 + 3 \cdot \alpha} \cdot \frac{3 + \beta}{5 + 4 \cdot \beta} \end{align} \]
\[ \begin{align}P(ball_1 = \mbox{Topic 2} | z_{-i}, W) \propto& \frac{0 + \alpha}{4 + 3 \cdot \alpha} \cdot \frac{1 + \beta}{5 + 4 \cdot \beta} \end{align} \]
\[ \begin{align}P(ball_1 = \mbox{Topic 3} | z_{-i}, W) \propto& \frac{2 + \alpha}{4 + 3 \cdot \alpha} \cdot \frac{0 + \beta}{5 + 4 \cdot \beta} \end{align} \]
Here, we won’t get into setting \(\alpha\) and \(\beta\), but for now assume that \(\alpha = 0.3\) and \(\beta = 0.25.\)
\[ \require{cancel} \begin{align}P(ball_1 = \mbox{Topic 1} | z_{-i}, W) \propto& \frac{2 + \cancel{\alpha} \overset{0.3}{}}{4 + 3 \cdot \cancel{\alpha} \overset{0.3}{}} \cdot \frac{3 + \cancel{\beta} \overset{0.25}{}}{5 + 4 \cdot \cancel{\beta} \overset{0.25}{}} = 0.2543 \end{align} \]
\[ \begin{align}P(ball_1 = \mbox{Topic 2} | z_{-i}, W) \propto& \frac{0 + \cancel{\alpha} \overset{0.3}{}}{4 + 3 \cdot \cancel{\alpha} \overset{0.3}{}} \cdot \frac{1 + \cancel{\beta} \overset{0.25}{}}{5 + 4 \cdot \cancel{\beta} \overset{0.25}{}} = 0.0128 \end{align} \]
\[ \begin{align}P(ball_1 = \mbox{Topic 3} | z_{-i}, W) \propto& \frac{2 + \cancel{\alpha} \overset{0.3}{}}{4 + 3 \cdot \cancel{\alpha} \overset{0.3}{}} \cdot \frac{0 + \cancel{\beta} \overset{0.25}{}}{5 + 4 \cdot \cancel{\beta} \overset{0.25}{}} = 0.0196 \end{align} \]
\(\rightarrow\) assign Topic 1 to the first instance of ball
Update the first instance of ball and move on to the second instance of ball. Keep iterating through the words until the words have converged and they aren’t changing topics any longer. (The LDA Gibbs sampler will converge, but only to a local optimum.)

If we have a document that has already been used to train the LDA, we know the topic allocations for each word in the document.
\[\mbox{Topic}(d) = \mbox{arg} \max_k \theta_{d,k}\]
where \(\theta_{d,k}\) is the probability of topic \(k\) for document \(d\).
\(\theta_{d,k}\) is estimated as the proportion of topic \(k\) in document \(d\).
For a new document, we do not have a topic allocation for each word in the document.
For each word in the document, you get a probability for each topic, based on the learned topic-word distribution
The probability of a topic \(k\) for the new document is essentially the weighted sum of the topic probabilities for all the words in the document, considering their relative importance.
The EM algorithm is an incredibly useful tool for solving complicated maximization procedures, particularly with respect to maximizing likelihoods (typically for parameter estimation). We will describe the procedure here in the context of estimating the parameters of a two-component mixture model.
Consider the Old Faithful geyser Yellowstone National Park, Wyoming, USA with the following histogram of data on waiting times between each eruption:

\[\begin{align} Y_1 &\sim N(\mu_1, \sigma_1^2)\\ Y_2 &\sim N(\mu_2, \sigma_2^2)\\ Y &= (1-\Delta) Y_1 + \Delta Y_2\\ P(\Delta=1) &= \pi\\ \end{align}\] In the simple two component case, we can see that the representation above indicates that first we generate a \(\Delta \in \{0,1\}\), and then, depending on the result, we generate either \(Y_1\) or \(Y_2\). The likelihood associated with the above setting is:
\[\begin{align} g_Y(y) = (1-\pi) \phi_{\theta_1}(y) + \pi \phi_{\theta_2}(y) \end{align}\] where \(\phi_\theta\) represents the normal distribution with a vector \(\theta=(\mu, \sigma)\) of parameters. Typically, in statistical theory, to find \(\theta\), we would take the derivative of the log-likelihood to find the values which maximize. Here, however, the likelihood is too complicated to solve for \(\theta\) in closed form.
\[\begin{align} l(\theta; {\bf y}) = \sum_{i=1}^N \log [(1-\pi) \phi_{\theta_1}(y) + \pi \phi_{\theta_2}(y)]. \end{align}\]
If we know which point comes from which distribution, however, the maximization is straightforward in that we can use the points in group one to estimate the parameters from the first distribution, and the points in group two to estimate the parameters in the second distribution. The process of assigning points and estimating parameters can be thought of as two steps:
Algorithm: EM Algorithm for two-component Gaussian mixture. From The Elements of Statistical Learning (2001), by Hastie, Tibshirani, and Friedman, pg 238.
The algorithm shows that for a particular allocation of the points, we can maximize the given likelihood to estimate the parameter values (done in the Maximization Step). However, it is not obvious from the algorithm that the first allocation step leads to a maximization (local or global) of the likelihood. The proof of the EM algorithm converging to a local maximum likelihood (it does not necessarily converge to a global max) uses information on the marginal prior and posterior likelihoods of the parameter values and Jensen’s inequality to show that the likelihood does not decrease through the iterative steps.
Note that in the previous \(k\)-means algorithm we iterated between two steps of assigning points to clusters and estimating the cluster centers (we thought of the space as scaled so that the Euclidean distance was appropriate in all dimensions). Two differences in the algorithms we covered are:
Indeed, although the EM-algorithm above is slightly different than the previous \(k\)-means algorithm, the two methods typically converge to the same result and are both considered to be different implementations of a \(k\)-means algorithm.
See the following applet for a visual representation of how the EM-algorithm converges: http://www.socr.ucla.edu/applets.dir/mixtureem.html.
Heavily inspired by Luis Serrano’s youtube video. Also note his other educational materials↩︎
